随着现代科技的飞速发展,新型电子产品以及新能源汽车的出现,人们对锂电池的容量,能量密度以及安全性提出了更高标准的要求。锂电池安全高效的运行离不开电解质的稳定运作,其基本功能是在电池的正负极之间作为内部电荷传递的主要介质,并防止电池发生内短路现象。理想的电解质应该促进电极表面的法拉第反应动力学过程,同时缓解锂电池界面处的副反应。传统的碳酸酯基电解液体系由于其较高的脱溶剂化能垒和较差的界面相容性,通常会导致电极界面缓慢的反应动力学和较大的副反应。
最近,浙江大学我院陈立新教授、范修林研究员团队提出了一种新的分子对接电解液(MDE)设计策略,该策略克服了锂盐在隐性溶剂分子(具有潜在的离子配位官能团,但是由于空间位阻等导致无法溶剂化锂离子)中解离的局限性,同时在电极界面处实现了快速稳定的电化学反应。
该项研究成果于北京时间2024年7月15日,发表在国际顶级期刊《Nature Chemistry》。论文第一作者为浙江大学马宝琛和张海阔博士研究生,通讯作者为浙江大学范修林研究员,并受到腾讯优图实验室,浙江大学陈立新教授、肖学章副教授和马里兰大学邓涛博士(现为上海交大中英低碳研究院副教授)的大力支持。浙江大学为该论文的唯一通讯单位。
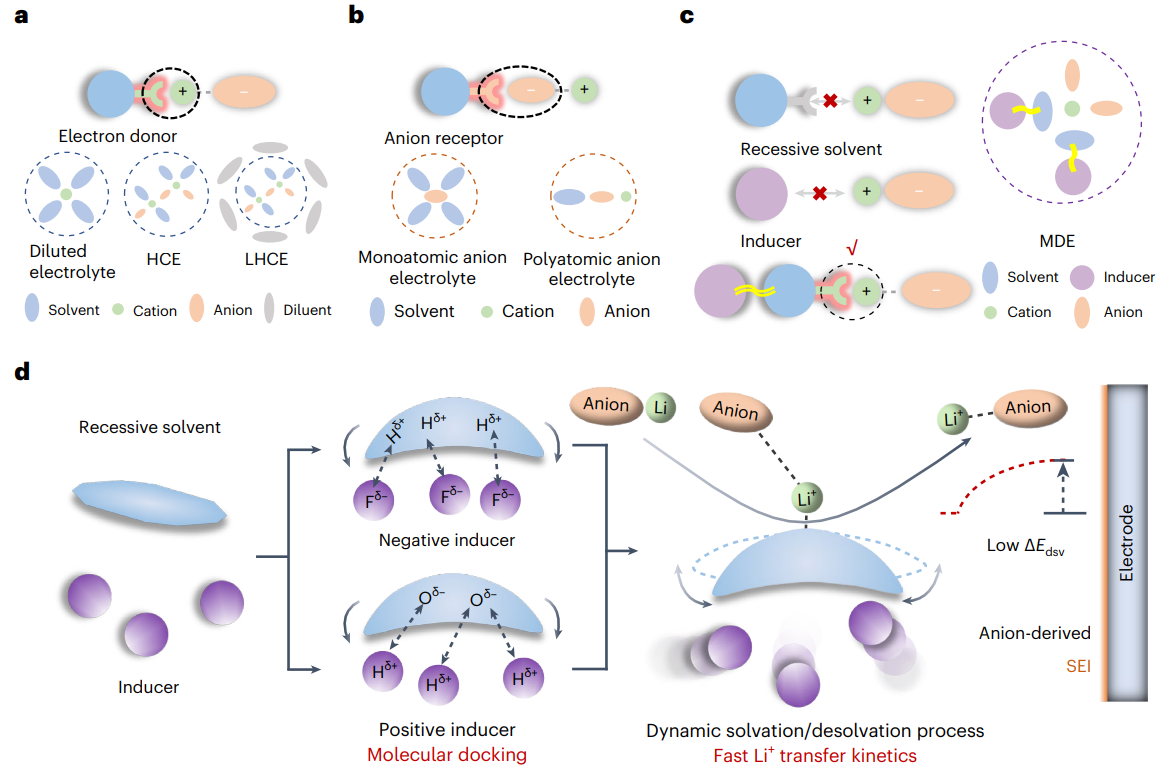
图1 目前的电解液设计所用的显性溶剂化(a,b)与本文提出的隐性溶剂化模型(c,d)示意图
传统电解液体系目前主要基于两种机制:电子供体或阴离子受体。根据其特定的溶剂化结构,可进一步分为稀电解液、高浓电解液、局部高浓电解液、单原子阴离子电解液以及多原子阴离子电解液等。以上电解液体系都是通过自发的离子-溶剂配位形成的,称为显性溶剂化。然而,在隐性溶剂化模型中,单独的隐性溶剂或诱导剂不能溶解锂盐,自然也不具备离子电导率支撑电池的正常运行。当隐性溶剂与某些特定的氟苯类或卤代烷类化合物(称为诱导剂)混合时,诱导剂分子会通过非典型氢键相互作用(特别是Fδ--Hδ+或Hδ+-Oδ-相互作用)使得隐性溶剂分子产生构型变化,影响分子表面静电势能分布,进而激活隐性溶剂分子的Li+配位点。这些分子间的动态相互作用形成了Li+-溶剂间的动态配位过程,降低了Li+界面处的脱溶剂化能垒,同时提高了阴离子界面还原动力学,缓解了电解液溶剂分子在正/负极侧的副反应。同时,诱导剂调控的离子动态脱溶剂化过程也使得Li+-FSI+离子对配位能力增强,从而在电极侧形成阴离子衍生的SEI。
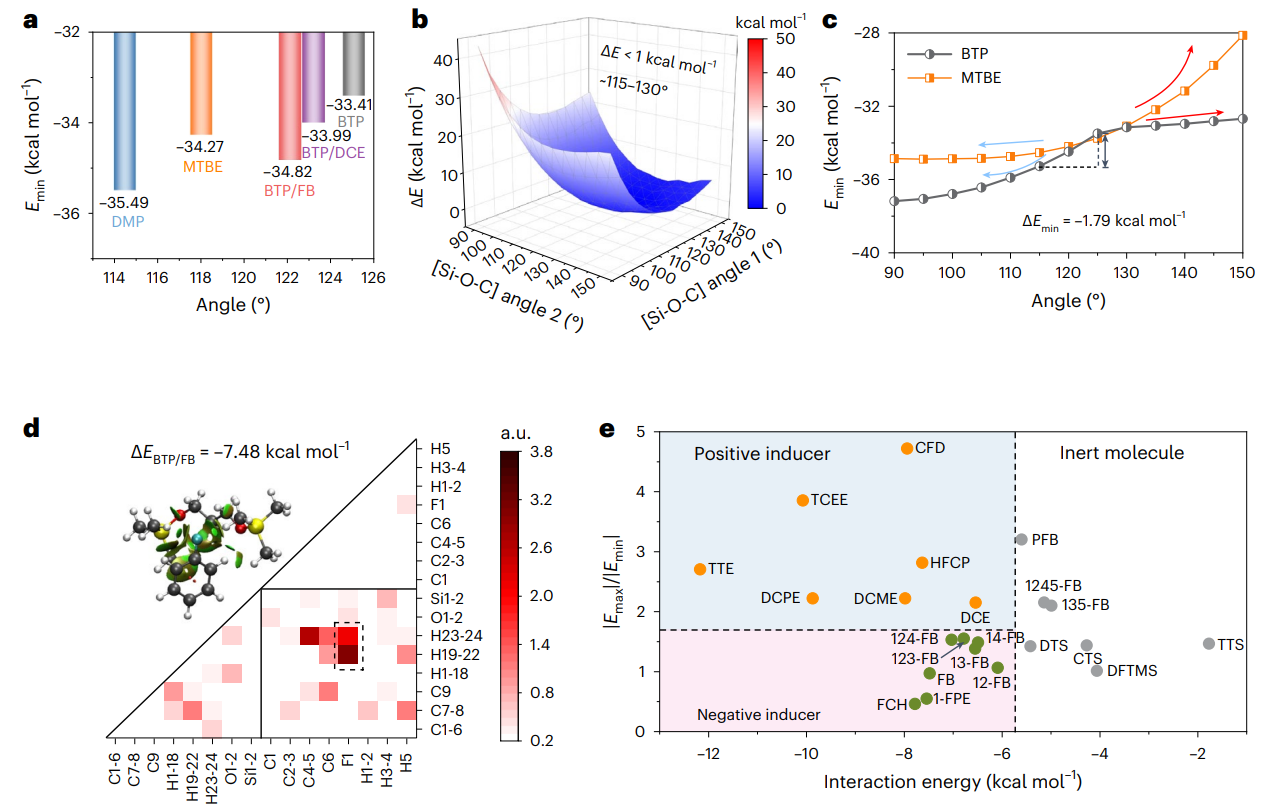
图2 隐性溶剂与诱导剂相关计算模拟
为了全面的解析分子对接机制和结构-性能的对应关系,选择1,3-双(三甲基硅氧基)丙烷(BTP)以及1,2-双(三甲基硅氧基)乙烷(BTE)作为典型的隐性溶剂分子,基于自然键轨道以及分子静电势能分析,揭示了隐性溶剂分子构型、空间位阻、键角大小等与离子配位能力的内在关系,提出诱导剂作用力阈值(< 5.7 kcal/mol)并结合实验结果,筛选并设计出了15种氟苯、卤代烷烃类诱导剂分子。结合核磁、红外光谱、拉曼、等温量热滴定法、飞秒瞬态吸收光谱等测试表征手段进一步证明了隐性溶剂分子与诱导剂之间的分子对接机制。
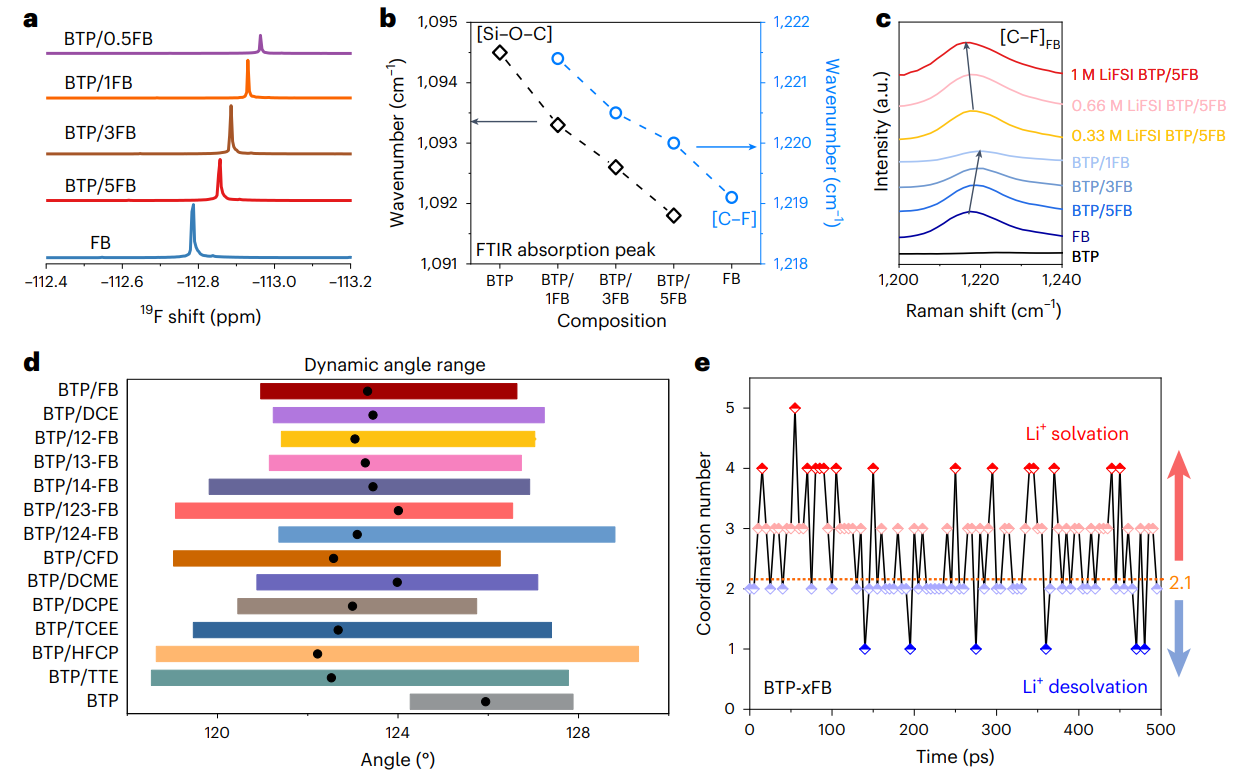
图3 分子对接电解液测试表征与模拟计算
通过密度泛函理论(DFT)与分子动力学(MD)模拟发现,相较于纯隐性溶剂BTP分子,诱导剂的引入会使得隐性溶剂分子[Si-O-C]键角向小角度压缩,同时分子中O位点静电势能减小,对锂离子的配位能力增强。与此同时,由于诱导剂分子在隐性溶剂分子周围的热运动,诱导剂的配位数会出现明显的随机波动,高配位数(≥3)的诱导剂有利于Li+的溶剂化,低配位数(1或2)的诱导剂有利于Li+自发脱离溶剂化壳结构。因此,MDE通过诱导剂与隐性溶剂分子间的动态对接/解耦过程,导致Li+-溶剂相互作用的动态溶剂化/脱溶剂化过程,有助于降低Li+脱溶剂化能垒,实现快速的Li+界面电化学反应。
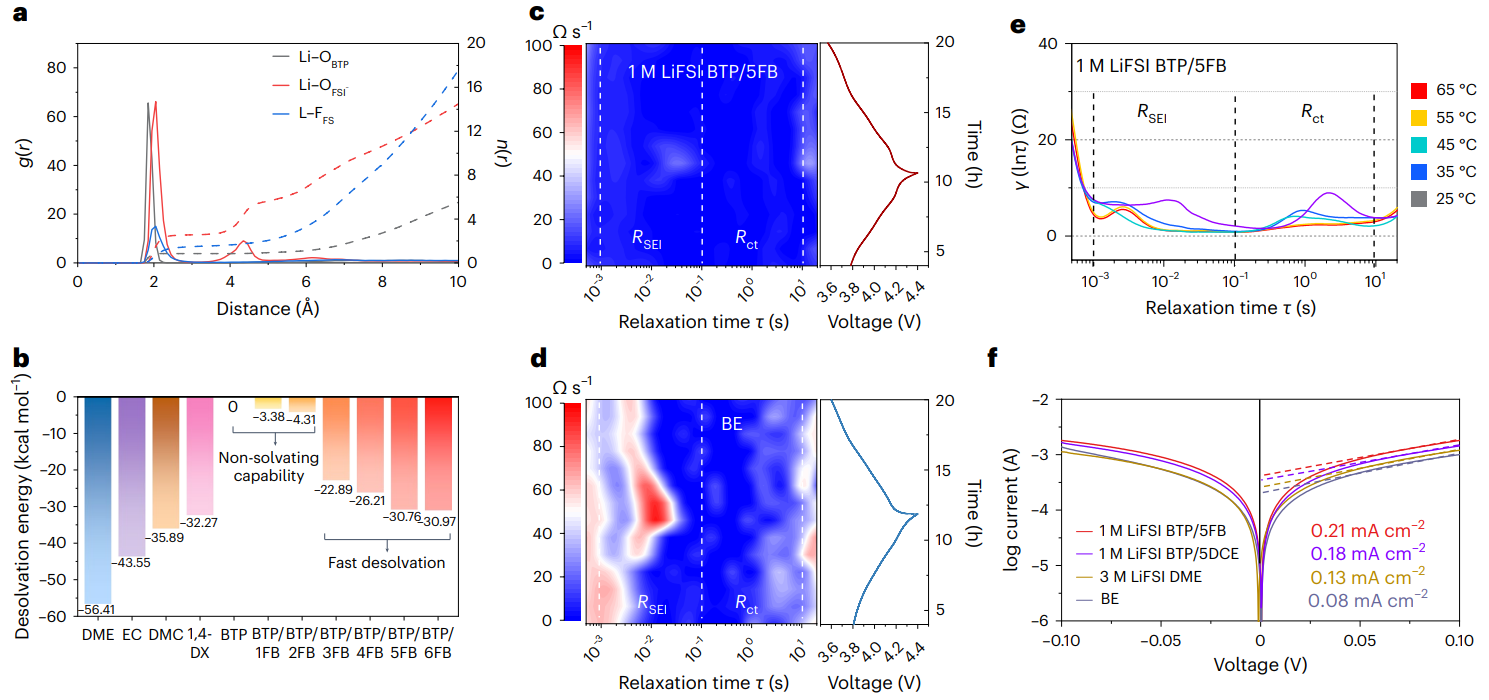
图4 动态锂离子溶剂配位实现快速的传输动力学
进一步的DFT、MD计算模拟以及弛豫时间分析实验证明了相较于传统的酯基电解液,MDE中较低的离子脱溶剂化能垒以及生成低界面阻抗的SEI,有利于提高MDE对于锂金属界面的兼容性。
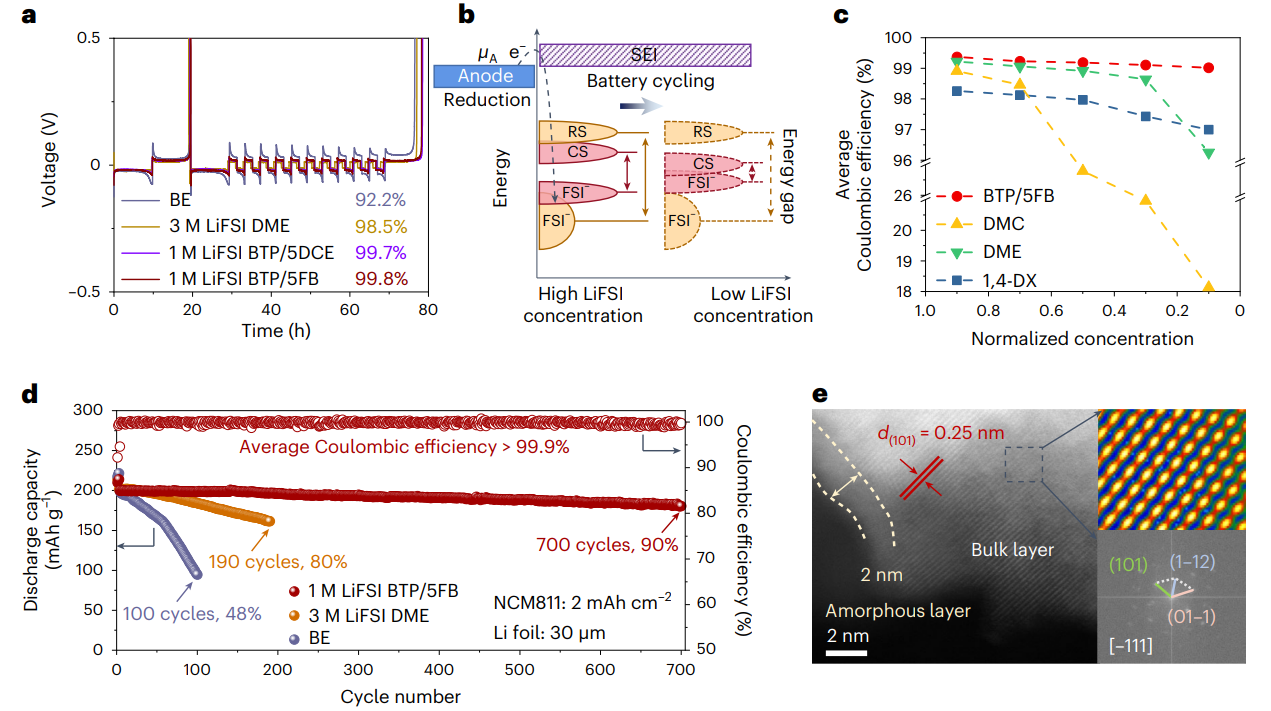
图5 锂金属负极以及全电池循环性能分析
基于分子对接电解液的设计策略,电解液的锂铜库伦效率可超过99.5%,在4.4 V 30 μm Li||2.0 mAh cm-2 LiNi0.8Co0.1Mn0.1O2(NCM811)全电池中,700次循环后的容量保持率为90%。在1 Ah 3.4 mAh cm-2石墨||3.0 mAh cm-2 NCM811软包电池中,550次循环后的容量保持率为98%。同时该策略设计的电解液对锂离子电池体系也具有良好的适配性。
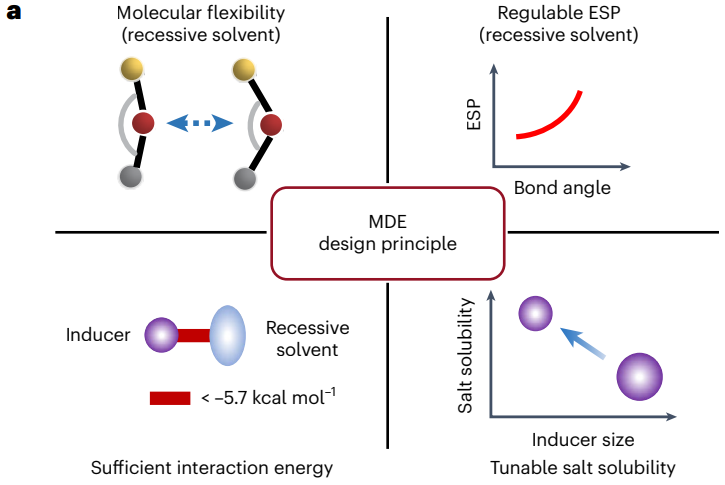

图6 分子对接电解液的设计原则和相关指标评价
在这项工作中,作者提出了一种基于分子对接机制的新型电解液设计策略,有望提升锂离子/金属电池中界面动力学和同时抑制界面副反应,实现锂电池的高效稳定循环。设计MDE的关键因素包括隐性溶剂的独特分子柔性(例如,BTP硅氧烷分子的绝热弯曲势能< 1.7 kcal/mol),同时分子键角的变化对应着分子静电势能的调节(例如在分子键角减小时溶剂配位官能团的静电势较低),以及非典型氢键(Fδ--Hδ+或Hδ+-Oδ-相互作用)引起的足够的分子间相互作用(< -5.7 kcal/mol)。此外,使用更小尺寸的诱导剂,减小分子对接过程中的位阻效应,可以进一步提升锂盐在隐性溶剂中的溶解度。
